よくあるご質問
近接ライゲーションテクノロジーDovetail Genomics®
すべて開く
(Dovetail® Omni-C® アッセイ & キット) 制限酵素を使用せずに有効なリードをどのようにコールできるのでしょうか?
制限酵素と超音波処理の使用により、従来のHi-Cの有効なリードは、リードペアの挿入サイズ(または物理的カバレッジ)がライブラリ断片よりも大きく、かつ制限酵素サイトに隣接している必要があります。Micro-Cライブラリは制限酵素を使用せず、超音波処理も必要としないため、有効なMicro-Cリードは、リードペアの挿入サイズ(または物理的カバレッジ)がライブラリ断片よりも大きいことのみが要件となります。詳細については、こちらのホワイトペーパーをご確認ください。
(Dovetail® Omni-C® アッセイ & キット) ワークフローを完了するまでにどのくらいの時間がかかりますか?
サンプルからシークエンス可能なライブラリを作成するまで、2日間です。
(Dovetail® 近接ライゲーションアッセイ) HiChIP、Micro-C、Omni-Cとは何ですか?
これらはDovetail Genomics®のみが提供する新しいHi-Cアッセイで、伝統的なHi-Cとは以下の重要な点で異なります。
HiChIPは、ChIP-seqのクロマチン免疫沈降とHi-Cのコンフォメーションキャプチャを組み合わせることで、クロマチンのフォールディングについてタンパク質指向型の視点を提供します。
Micro-Cは、制限酵素の代わりにMNaseを使用するHi-Cで、Hi-Cアッセイの中で最も高い解像度を実現し、ヌクレオソームレベルでのフォールディングを解析可能です。
Omni-Cは、制限酵素の代わりにDNaseを使用するHi-Cで、均一なゲノムカバレッジを生成します。
上記のアッセイのうち、最適なアプローチをご提案させていただきますので、ぜひご相談ください。
(Dovetail® 近接ライゲーションアッセイ) Hi-Cについて全くの初心者で、これまでに一度も使用したことがありませんが、キットを使うことができますか?
Dovetailの近接ライゲーションキットはユーザーフレンドリーで、Hi-C初心者向けに設計されています。さらに、Dovetailの世界トップクラスのサポートチームも、問題が発生した場合や質問がある場合にサポートを提供しています。
(Dovetail® 近接ライゲーションアッセイ) Dovetailではキットや受託解析サービスを提供していますか?
両方を提供しています。キットと受託解析サービスの詳細についてはお問い合わせください。
(Dovetail® 近接ライゲーションアッセイ) クロマチンコンフォメーションキャプチャ、または Hi-C とは何でしょうか?
クロマチンコンフォメーションキャプチャ(Chromosome conformation capture, 3C)は、近接ライゲーションとも呼ばれ、クロマチンの空間的構成を解析するために設計された分子生物学的手法です。ライゲーションイベントにビオチンをタグ付けし、ストレプトアビジンで分離してペアエンド全ゲノムシークエンスを行う手法は、Hi-C と呼ばれています。
詳細については、Davis et al. (2017)のレビュー論文をご参照ください。
https://www.nature.com/articles/nmeth.4146
(Dovetail® Omni-C® アッセイ & キット) 解析にはどれぐらいのシークエンスデプスとライブラリ数が必要ですか?
これは主にアプリケーションによって異なります。以下の推奨例をご参照ください。
ゲノムアセンブリ; 3 Gbp 未満の場合、30x カバレッジ、1 ライブラリ
ジェノタイピングとハプロタイプフェーシング; 40x カバレッジ、2 ライブラリ
トポロジー解析; A/Bコンパートメント(3x カバレッジ、1 ライブラリ)、TADs(30x、1~2 ライブラリ)、Loop(> 90x、3~4 ライブラリ)
QCには100~200万ペアエンドリード(2 × 150 bp)を推奨します。
(Dovetail® Omni-C® アッセイ & キット) Omni-Cデータは、従来の制限酵素ベースのHi-Cデータで使用されるオープンソースツールと互換性がありますか?
HiC-ProはOmni-Cライブラリとも互換性があります。ただし、必要なカスタム設定ファイルが存在します。HiC-Proの実行方法に関するガイダンスが必要な場合はお問い合わせください。
(Dovetail® Omni-C® アッセイ & キット) 解析を開始するために必要なファイル形式は何ですか?
DovetailツールによるQCとコンタクトマトリックスの生成には、Omni-Cのrawシーケンスデータ(*.fq.gz)とリファレンスゲノムアセンブリ(*.fa)が必要です。
(Dovetail® Omni-C® アッセイ & キット) Dovetailではドライ解析のサポートをしていますか?
Dovetailは、完全なドキュメント付きで包括的なベストプラクティスのステップバイステップワークフローのご用意があります。
https://omni-c.readthedocs.io/en/latest/
アラインメント、ペア生成、高品質な近接ライゲーションリードのフィルタリング、.hicファイルと.coolファイルの両方に対応したコンタクトマトリックスの生成、その他、一般的に使用されるアプリケーションをカバーしています。
(Dovetail® Omni-C® アッセイ & キット) ライブラリをどのようにシーケンスすべきですか?
Omni-C ライブラリは Illumina 互換です。2 x 150 bp のシークエンスを推奨します。通常は1 つのライブラリで ~300M リードペアで十分ですが、最終的なシークエンスデプスに応じて、シークエンスの実行前に複数のライブラリをプールする必要がある場合があります。
(Dovetail® Omni-C® アッセイ & キット) 高品質なOmni-Cライブラリを作成するためにどのようなQCが必要ですか?
Omni-Cのワークフローには、消化反応の評価用などのQCステップが3段階で組み込まれています。また、作成されたライブラリの最終的なQCとして、ディープシークエンスを実施する前に浅めのシークエンスを実施してQCを行うことを推奨しています。キットのユーザーは、使いやすいQC解析パイプラインを利用可能です。
(Dovetail® Omni-C® アッセイ & キット) ライブラリを作成するためにキット以外に必要な機器は何ですか?
0.2 mLと1.5 mLチューブ用の磁気分離ラック、遠心機、スイングバケットローター、ボルテックスミキサー、サーマルサイクラー、サーマルミキサー、Qubit、TapeStation(またはFragment Analyzer、Bioanalyzer)が必要になります。
(Dovetail® Omni-C® アッセイ & キット) 超音波処理はどのようなものが推奨ですか?
Omni-Cワークフローでは断片化は酵素で行われるため、超音波処理は不要です。
(Dovetail® Omni-C® アッセイ & キット) Omni-Cアッセイは他社製のライブラリ調製キットと互換性がありますか?
Dovetailは、ライブラリ調製ワークフローを効率化するためのライブラリ調製モジュールとインデックスプライマーモジュールを提供していますが、Omni-Cでは、他社製のライブラリ調製キットとも互換性があります。NEBNext® Ultra™ II DNAライブラリ調製キット、Kapa® HyperPrep、Swift Biosciences Accel-NGS® 2S Plus DNAライブラリキットが検証済みです。
(Dovetail® Omni-C® アッセイ & キット) 既にホルムアルデヒドでクロスリンクされた凍結細胞ペレットを保有しています。これらはOmni-Cアッセイのインプットとして用いることができますか?
ホルムアルデヒド濃度≥1%で事前にクロスリンクされた凍結細胞であれば、アッセイのインプットとして使用可能です。ユーザガイドの2段階固定化プロセスのホルムアルデヒド固定化ステップを省略してください。
(Dovetail® Omni-C® アッセイ & キット) ハイブリッドキャプチャーを実施する場合、プローブを設計する際にはどのような点を考慮する必要がありますか?
Omni-Cライブラリはゲノム全体をWGS(全ゲノムシーケンス)のようなカバレッジでカバーし、制限酵素を使用しません。そのため、市販のプローブセットを使用するか、制限酵素サイトを含まない独自のプローブを設計することができます。
(Dovetail® Omni-C® アッセイ & キット) Omni-C キットは、Capture-C などのハイブリッドキャプチャーアプローチと互換性がありますか?
Omni-C キットはハイブリッドキャプチャーアプローチと互換性があります。Omni-C ワークフローに、Agilent SureSelect、Illumina TruSight、IDT xGEN を統合可能です。
(Dovetail® Omni-C® アッセイ & キット) キットには何反応分が含まれていますか?
Omni-Cキット1つにつき、8反応分が含まれています。
(Dovetail® Omni-C® アッセイ & キット) 推奨されるサンプルの種類とサンプルの前処理方法はどのようなものですか?

(Dovetail® Omni-C® アッセイ & キット) Omni-Cは、ATリッチや反復配列の多いゲノムなどでも機能しますか?
Omni-Cのケミストリーは堅牢性が高く、多様なゲノムタイプに対応しています。
(Dovetail® Omni-C® アッセイ & キット) Dovetail Omni-Cキットで対応可能なサンプルの種類は何ですか?
細胞、動物や植物の組織、血液など、様々なサンプルに対応が可能です。
(Dovetail® Omni-C® アッセイ & キット) Dovetail Omni-C アッセイに必要な細胞のインプット量はどれくらいですか?
標準的な細胞インプット量は100万細胞です。
(Dovetail® Omni-C® アッセイ & キット) Dovetail Omni-C アッセイは Hi-C とどのように異なるのでしょうか?
Omni-Cは、制限酵素ではなくDNaseを用いてクロマチンを分解します。このアプローチはゲノム全体にわたる優れたカバレッジを提供し、ジェノタイピングやハプロタイプフェーシングなど、より多くの応用が可能になります。
(Dovetail® HiChIP MNase アッセイ & キット) 制限酵素を使用せずに有効なリードをどのようにコールできるのでしょうか?
制限酵素と超音波処理の使用により、従来のHi-Cの有効なリードは、リードペアの挿入サイズ(または物理的カバレッジ)がライブラリ断片よりも大きく、かつ制限酵素サイトに隣接している必要があります。Dovetail HiChIPライブラリは制限酵素を使用せず、超音波処理も必要としないため、有効なDovetail HiChIPリードは、リードペアの挿入サイズ(または物理的カバレッジ)がライブラリ断片よりも大きいことのみが要件となります。詳細については、こちらのホワイトペーパーをご確認ください。
(Dovetail® HiChIP MNase アッセイ & キット) 解析を開始するために必要なファイル形式は何ですか?
Dovetail HiChIP QCツールによるライブラリの評価には、HiChIPのrawシーケンスデータ(*.fq.gz)とリファレンスゲノムアセンブリ(*.fa)、1D ChIP-seqピーク位置のファイル(*.bed)が必要です。
(Dovetail® HiChIP MNase アッセイ & キット) Dovetailではドライ解析のサポートをしていますか?
Dovetailは、完全なドキュメント付きで包括的なベストプラクティスのステップバイステップワークフローのご用意があります。
https://hichip.readthedocs.io/en/latest/
アラインメント、ペア生成、高品質な近接ライゲーションリードのフィルタリング、IPエンリッチメントの評価、.hicファイルと.coolファイルの両方に対応したコンタクトマトリックスの生成、その他、一般的に使用されるアプリケーションをカバーしています。
(Dovetail® HiChIP MNase アッセイ & キット) Dovetail HiChIPライブラリにはどれぐらいのシークエンスデプスが必要ですか?
対象とするタンパク質とゲノムにおけるタンパク質-DNA相互作用の発生率によって異なります。以下の推奨される開始ガイドラインをご参照ください;
QC; 1 Mのペアエンドリード(2 x 150 bp)でライブラリの近接ライゲーションの品質を評価します。ChIPエンリッチメントの評価には20 M – 40 Mのペアエンドリード(2 x 150 bp)が必要です。
ディープシークエンス; 1つのHiChIPライブラリを約150 Mのリードペア(2 × 150 bp)でシークエンスすることを推奨します。
低頻度の場合; 100 Mのペアエンドリード(2 × 150 bp)、1ライブラリ。
高頻度の場合; 400 Mのペアエンドリード(2 × 150 bp)、2~4ライブラリ。
(Dovetail® HiChIP MNase アッセイ & キット) 高品質なHiChIPライブラリを作成するためにどのようなQCが必要ですか?
Dovetail HiChIP MNaseワークフローには、消化反応の評価用QCなどのステップが組み込まれています。また、作成されたライブラリの最終的なQCとして、ディープシークエンスを実施する前に浅めのシークエンスを実施してQCを行うことを推奨しています。キットのユーザーは、使いやすいQC解析ワークフローにアクセスすることが可能です。
(Dovetail® HiChIP MNase アッセイ & キット) Dovetail HiChIP MNaseアッセイを実施するにはどのくらいの時間がかかりますか?
サンプルからシークエンス可能なライブラリを作成するまで、わずか3日で完了します。
(Dovetail® HiChIP MNase アッセイ & キット) Dovetail HiChIP MNase アッセイで対応可能なサンプルの種類は何ですか?
Dovetail HiChIP MNase アッセイは現在、哺乳類細胞で検証済みです。他のサンプル種類にご興味がある場合はお問い合わせください。
(Dovetail® HiChIP MNase アッセイ & キット) Dovetail HiChIPに必要な細胞のインプット量はどれくらいですか?
対象の抗体/タンパク質によって異なります。以下の表をご参照ください。

(Dovetail® HiChIP MNase アッセイ & キット) Dovetail HiChIP MNase アッセイはヌクレオソームフリー領域をカバーしていますか?
ヌクレオソームフリー領域ではカバレッジが低下しますが、消化プロファイルには二量体ヌクレオソーム、三量体ヌクレオソーム、および単量体ヌクレオソームが含まれているため、カバレッジがゼロになることはほとんどありません。そのため、一部の断片にリンカーDNAが含まれ、ヌクレオソームフリー領域でもカバレッジが確保されています。
(Dovetail® HiChIP MNase アッセイ & キット) ChIP-seqデータは必要ですか?
ChIP-seqデータを組み込むことで、データのQCと解釈をサポートすることができますが、必須ではありません。ChIP-seqデータを生成していない場合に、ENCODEなどの公開データソースからデータを取得して使用することもできますが、ご自身の実験やサンプルについて完全に反映したものではなくなります。
(Dovetail® HiChIP MNase アッセイ & キット) Dovetail HiChIP アッセイは他の HiChIP プロトコルとどのように異なるのでしょうか?
Dovetail HiChIP MNase アッセイは、Micro-C(MNase ベースの Hi-C)ワークフローを活用してクロマチン消化を実施します。そのためHiChIP MNase アッセイでは;
・プロトコルに超音波処理が不要なため、アッセイの信頼性が向上します。
・ヌクレオソームの位置が保持されます
・制限酵素と目的DNA-タンパク質相互作用の近接性に基づくピークの解析時の調整が不要です。(制限酵素を使用するHiChIPでは、タンパク質-DNA相互作用に最も近い制限部位でライブラリをエンリッチしているため、データ解析時にピークを、目的の相互作用に最も近い制限部位ではなく、実際のタンパク質-DNA部位を反映するように調整する必要があります)
(Dovetail® Micro-C アッセイ & キット) Micro-Cライブラリにはどれぐらいのシークエンスデプスが必要ですか?
QCのためには、100~200万のリードペアを推奨します。
ディープシークエンスの場合、1つのMicro-Cライブラリを最大約300万リードペアまでシークエンスすることを推奨します。
トポロジー解析: A/Bコンパートメント(3xカバレッジ; 1ライブラリ)、TADs(30x; 1~2ライブラリ)、Loop(80xカバレッジ; 2~3ライブラリ)
(Dovetail® Micro-C アッセイ & キット) 制限酵素を使用せずに有効なリードをどのようにコールできるのでしょうか?
制限酵素と超音波処理の使用により、従来のHi-Cの有効なリードは、リードペアの挿入サイズ(または物理的カバレッジ)がライブラリ断片よりも大きく、かつ制限酵素サイトに隣接している必要があります。Micro-Cライブラリは制限酵素を使用せず、超音波処理も必要としないため、有効なMicro-Cリードは、リードペアの挿入サイズ(または物理的カバレッジ)がライブラリ断片よりも大きいことのみが要件となります。詳細については、こちらのホワイトペーパーをご確認ください。
(Dovetail® Micro-C アッセイ & キット) 解析を開始するために必要なファイル形式は何ですか?
DovetailツールによるQCとコンタクトマトリックスの生成には、Micro-Cのrawシーケンスデータ(*.fq.gz)とリファレンスゲノムアセンブリ(*.fa)が必要です。
(Dovetail® Micro-C アッセイ & キット) Dovetailではドライ解析のサポートをしていますか?
Dovetailは、完全なドキュメント付きで包括的なベストプラクティスのステップバイステップワークフローのご用意があります。
https://micro-c.readthedocs.io/en/latest/
アラインメント、ペア生成、高品質な近接ライゲーションリードのフィルタリング、.hicファイルと.coolファイルの両方に対応したコンタクトマトリックスの生成、その他、一般的に使用されるアプリケーションをカバーしています。
(Dovetail® Micro-C アッセイ & キット) Micro-Cライブラリをシークエンシングする際、どのリード長を選択すべきですか?
Micro-CライブラリはIllumina互換です。2 x 75 bp、2 x 100 bp、または2 x 150 bpのシークエンシングを推奨します。
(Dovetail® Micro-C アッセイ & キット) 実験デザインをどのようにアプローチすれば、下流の解析を可能にできますか?
実験デザインは、利用可能な下流の解析の種類を決定するため、データを生成する前に両者を同時に考慮することが重要です。Micro-Cの実験デザインに関する詳細な説明はこちらでご確認いただけます。
https://dovetailgenomics.com/wp-content/uploads/securepdfs/2022/03/Micro-C_Exp_Design.pdf
(Dovetail® Micro-C アッセイ & キット) 高品質なMicro-Cライブラリを作成するためにどのようなQCが必要ですか?
Micro-Cワークフローには、消化反応の評価用QCなどのステップが組み込まれています。また、作成されたライブラリの最終的なQCとして、ディープシークエンスを実施する前に浅めのシークエンスを実施してQCを行うことを推奨しています。キットのユーザーは、使いやすいQC解析ワークフローにアクセスすることが可能です。
(Dovetail® Micro-C アッセイ & キット) ライブラリを作成するためにキット以外に必要な機器は何ですか?
0.2 mLと1.5 mLチューブ用の磁気分離ラック、遠心機、スイングバケットローター、ボルテックスミキサー、サーマルサイクラー、サーマルミキサー、Qubit、TapeStation(またはFragment Analyzer、Bioanalyzer)が必要になります。
(Dovetail® Micro-C アッセイ & キット) 超音波処理はどのようなものが推奨ですか?
Micro-Cワークフローでは断片化は酵素で行われるため、超音波処理は不要です。
(Dovetail® Micro-C アッセイ & キット) キットのワークフローを完了するまでにどのくらいの時間がかかりますか?
サンプルからシークエンス可能なライブラリを作成するまで、1.5日しかかかりません。
(Dovetail® Micro-C アッセイ & キット) キットには何反応分が含まれていますか?
Micro-Cキット1つにつき、8反応分が含まれています。
(Dovetail® Micro-C アッセイ & キット) Dovetail Micro-Cキットで対応可能なサンプルの種類は何ですか?
現在、Micro-Cは哺乳類細胞、哺乳類組織、哺乳類の凍結保存PBMC、および新鮮な哺乳類全血で検証済みです。また、ユーザー開発のショウジョウバエとコメのプロトコルもあります。他のサンプル種類にご興味がある場合はお問い合わせください。
(Dovetail® Micro-C アッセイ & キット) Dovetail Micro-C アッセイに必要な細胞のインプット量はどれくらいですか?
Micro-Cアッセイの標準的な細胞インプット量は100万細胞です。
(Dovetail® Micro-C アッセイ & キット) Micro-Cのデータはヌクレオソームフリー領域をカバーしていますか?
ヌクレオソームフリー領域ではカバレッジが低下しますが、消化プロファイルには二量体ヌクレオソーム、三量体ヌクレオソーム、および単量体ヌクレオソームが含まれているため、カバレッジがゼロになることはほとんどありません。そのため、一部の断片にリンカーDNAが含まれ、ヌクレオソームフリー領域でもカバレッジが確保されています。
(Dovetail® Micro-C アッセイ & キット)Dovetail Micro-C アッセイは Hi-C とどのように異なるのでしょうか?
Micro-Cは、通常のHi-Cが使用する制限酵素の代わりに、マイクロコッカスヌクレアーゼ(MNase)を用いてクロマチンを消化します。このアプローチは優れたシグナル/ノイズ比によりヌクレオソームの分解能/ヌクレオソームのフェージング解析を可能にし、エンハンサー-プロモーター/プロモーター-プロモーター相互作用や、Loopなど3次元クロマチン構造の詳細をより高精度に視覚化することができます。
(Dovetail® LinkPrep™ アッセイ & キット)LinkPrep™ のデータからどのようにして体細胞変異を検出しますか?
標準的な NGS ツールを使用してデータを解析することが可能です。
Dovetail Genomicsでは、以下の readthedocs ページのご用意の他、
https://varilink.readthedocs.io/en/latest/
解析ポータル(有料)や、受託解析サービス(有料)も承っています。
(Dovetail® LinkPrep™ アッセイ & キット)どれぐらいのシークエンスデプスが推奨されますか?
QCシークエンスの場合は、100~1000万リードペア(2×150 bp)の浅いシークエンスで十分です。
標準的なシークエンスの場合は、1ライブラリにつき、最大3億リードペア(2×150 bp)までのシークエンスを推奨します。デプスは目的のバリアントの種類、腫瘍画分、バリアントアレル頻度によって異なります。

低VAFイベントの検出はシークエンスデプスに依存します。
(Dovetail® LinkPrep™ アッセイ & キット)なぜ遠心分離にスイングバケットローターが使用されるのでしょうか?
ペレットを可視化しやすくして細胞の損失を軽減するとともに、低速回転で細胞の凝集を軽減することで、タグメンテーション反応の効率低下を防ぐことができるためです。
(Dovetail® LinkPrep™ アッセイ & キット)他社製のライブラリ調製キットを使用できますか?
いいえ、LinkPrep™ キットは他社製のライブラリ調製キットの使用をサポートしていません。キットに構成されているモジュールをお使いください。
(Dovetail® LinkPrep™ アッセイ & キット)組織サンプルはどのように採取し、準備すればよいですか?
採取した組織を液体窒素で瞬間凍結し、使用するまで-80℃で保存してください。
(Dovetail® LinkPrep™ アッセイ & キット)Dovetail LinkPrep™ キットで100万個以下の細胞から実験を開始することは可能ですか?
細胞10万個まではプロトコルを変更せずに行うことができます。細胞数が少なくなるに従って、近接ライゲーション後の回収量が低下し、ライブラリの複雑さが低下する可能性があることにご注意ください。
(ゲノムアセンブリ受託サービス) Dovetail 受託サービスはどのような内容ですか?
「デノボアセンブリサービス」は、サンプルをお送りいただくだけで、ドラフトアセンブリ作成、近接ライゲーションライブラリ作成、スキャフォルディングまでを一括して行うことができます。「スキャフォルディングサービス」は、お手持ちのドラフトアセンブリとサンプルをお送りいただくことで、近接ライゲーションライブラリ作成以降の解析を行なうことができます。
(ゲノムアセンブリ受託サービス) Dovetail 受託サービスを利用することができない生物種はありますか?
生物種によっては、日本からDovetail社のあるアメリカまでの輸出・輸入が禁止、または制限がある場合があります。日本から輸出が可能か、アメリカに輸入が可能かを確認いたしますので、サンプルの生物種(学名)をお知らせください。この確認には数週間かかる場合があるため、あらかじめご了承ください。
(ゲノムアセンブリ受託サービス) Dovetail 倍数体にも対応できますか?
PacBio HiFi ロングリードとOmni-Cテクノロジーを組み合わせることで、多倍体のゲノムアセンブリにも対応が可能です。
サンプルにより、サブゲノムまたはハプロタイプの分解も可能ですのでご相談ください。
(ゲノムアセンブリ受託サービス) Dovetail サンプルはどのように輸送されますか?
国内輸送(お客様からトミーデジタルバイオロジー社まで)は、冷凍便でお送りください。弊社からDovetail社までは、ワールド・クウリアー社のサービスを使い冷凍輸送します。
(ゲノムアセンブリ受託サービス) Dovetail どれくらいの量のサンプルが必要ですか?
サンプルの生物種によって異なりますが、大まかな目安は以下の通りです。
動物 : 脳や肝臓など細胞密度の高い部位から、新鮮なうちに急速冷凍されたもの、200 mg程度。
植物 : 新芽や実生などできるだけ若い部位を、新鮮なうちに急速冷凍したもの、3 g 程度。
節足動物 : 胚や孵化したばかりの幼虫、初期の蛹などを、新鮮なうちに急速冷凍したもの、200 mg程度。
無脊椎動物 : 内臓(大型の動物の場合)、孵化したばかりの幼虫、精子、血液などを、新鮮なうちに急速冷凍したもの、500 mg程度。
サンプルのご準備に関する詳細は、別途資料がございますのでご連絡ください。
(ゲノムアセンブリ受託サービス) Dovetail QCはどのように行われますか?
Dovetail社で実施するクオリティチェックには以下のようなものがあり、お客様はチェックポイントごとの報告を受けることがあります。
1. ドラフトアセンブリ QC: ご提供いただいたドラフトアセンブリがHiRiseを使ったスキャフォルディングに使用できるかどうかをチェックします。
2. DNA QC : サンプルから抽出されたDNAの質、量、濃度がライブラリを作成するのに十分かどうかをチェックします。
3. ライブラリ QC : 近接ライゲーションライブラリが作成されたあと、少量のシークエンス(200M PE)を行ない、ドラフトアセンブリに対してマッピングします。このマッピング・アライメント情報を使用し、ライブラリの複雑性、リードペアの距離の分布、シグナルノイズ比を精査します。その結果、次に行なうディープシークエンスで、ゲノムカバレッジ 50x以上のデータを出力できると推測された場合、このクオリティチェックは通過となり、次のディープシークエンスに進みます。
4. スキャフォルドアセンブリ QC : HiRise パイプラインを使ってスキャフォルド解析を行った後、これまでの過去の結果を照らし合わせて、今回のアセンブリ結果が期待される結果だったかどうかが最終チェックされます。これが、お客様の元に納品される前の最後のQCです。
(ゲノムアセンブリ受託サービス) Dovetail データはどのように納品されますか?
基本的にはFTPによるダウンロードによりご提供いたします。ご要望に応じて、ハードディスクなどでデータを納品することも可能です。
ゲノムアセンブリ/スキャフォルディングサービス
・HiRiseによるゲノムアセンブリ結果(.fasta)
・概要レポート
・ドラフトスキャフォルドに推定されるミスジョイント情報
・ドラフトアセンブリ/スキャフォルドの位置情報
・アラインメントファイル(.bam)
・Omni-Cライブラリシークエンスデータ(.fasta)
+ TAD解析(可能な場合)
・コンタクトマトリクスファイル(.mcool, .hic)
・TADコール、CTCF結合サイト、A/Bコンパートメント、アイソコア位置(.bedpe)
・HiGlass用データと使用ガイド(.multires)
・概要レポート
エピジェネティクスサービス
・ライブラリシークエンスデータ(.fasta)
・アラインメントファイル(.bam)
・コンタクトマトリクスファイル(.mcool, .hic)
・ペアファイル(.pair)
・概要レポート
(ゲノムアセンブリ受託サービス) Dovetail 受託サービスではドラフトアセンブリはどのように作成しますか?
現在DovetailではPacBio HiFiロングリード × Hifiasmアセンブリの組み合わせを採用しています。
(ゲノムアセンブリ受託サービス) Dovetail 自分でドラフトアセンブリを作成する場合に、必要なデータ量はどれくらいですか?
illuminaの場合、目安として、2x150bpのペアエンドで最低60x カバレッジ、2x100bpの場合は90xカバレッジのデータ量が必要となります。
(ゲノムアセンブリ受託サービス) Dovetail HiRiseパイプラインに用いるドラフトアセンブリは、特別な方法で用意する必要がありますか?
いいえ。ドラフトアセンブリに使用するツールには、例えば、Discovar De Novo, SGA, Meraculous, Falconなど、様々なアセンブラがありますが、これでなければいけないという制限はありません。また、シークエンサーについても、HiRiseの必要条件(N50>100kb)に合致すれば、illuminaやPacBioなど制限はありません。しかし、最初のドラフトアセンブリの正確さが、次のスキャフォルディングの完成度に影響し、ひいてはアセンブリ全体の精度にも影響しますので、最初のドラフトアセンブリは、正確であればあるほど望ましいものになります。
(ゲノムアセンブリ受託サービス) Dovetail HiRiseに用いるドラフトアセンブリの必要最低条件は?
絶対的な必要最低条件ではありませんが目安として、
1)スキャフォルドまたはコンティグのN50が100kb以上
2)フローサイトメトリーなどで確認した推定ゲノムサイズの少なくとも75%がドラフトアセンブリに含まれていること
を推奨しています。より精度が高く、断片化の少ないドラフトアセンブリから、最終的には良いゲノムアセンブリ結果が得られるのはもちろんですが、これらはひとつの目安であり、サンプルのゲノムの特徴によって大きく変わります。
(ゲノムアセンブリ受託サービス) Dovetail N50は長いがエラーが多いドラフトアセンブリと、N50は短いがエラーが少ないドラフトアセンブリがある場合、どちらを解析に用いますか?
HiRiseの必要条件(N50>100kb)に合致する限り、N50は短くともエラーが少ないドラフトアセンブリを用いた方が、最終的には良いゲノムアセンブリ結果を得ることができます。
(ゲノムアセンブリ受託サービス) Dovetail 受託サービスで近接ライゲーションライブラリはどのように作成されますか?
Dovetail社では現在、近接ライゲーションライブラリの作成に通常、Omni-Cテクノロジーを用いています。サンプルのゲノム3Gbあたり1つの近接ライゲーションライブラリを作成します。
(ゲノムアセンブリ受託サービス) Dovetail 近接ライゲーションライブラリはどのようにシークエンスされますか?
Dovetail社では、HiSeq X シークエンサーを使用しています。
(ゲノムアセンブリ受託サービス) Dovetail 近接ライゲーションライブラリをシークエンスするために必要な条件は?
ペアエンドシークエンスで、最低100 bpあることが、HiRiseによる解析には必要です。より長いリード長(例:2x250bp)であれば、ハプロタイプの検出には効果的ですが、スキャフォルドの長さや質に大きく貢献するわけではありません。
(ゲノムアセンブリ受託サービス) Dovetail HiRiseアセンブリに必要な近接ライゲーションライブラリデータの条件は?
サンプルの生物種によって異なります。これに影響を及ぼす要因として以下のものがあります。
・ゲノム :サイズ、リピート配列の割合、GC含量、ヘテロ接合性
・近接ライゲーションライブラリ :複雑性、シグナルノイズ比、リードペアの距離分布(インプットDNAのサイズ)
・ドラフトアセンブリ : コンティグの長さと精度
ゲノムアセンブリに必要な近接ライゲーションライブラリのシークエンスデータは、ゲノム全体に均一に30x以上のカバレッジが必要です。
(ゲノムアセンブリ受託サービス) Dovetail 近接ライゲーションライブラリのデータは、たくさんあればあるほど、アセンブリ結果が良くなるものですか?
サンプルに依存しますが、多くとも100xカバレッジで十分な結果を得ることができます。Dovetail社では現在も、近接ライゲーションライブラリのデータ量がアセンブリの結果にどう影響するか調査していますので、これ以上データを増やして改善する場合もあるかもしれません。
(ゲノムアセンブリ受託サービス) Dovetail HiRiseでのスキャフォルドに必要なデータは何ですか?
HiRise パイプラインへは、ドラフトアセンブリ(N50>100kb)と、近接ライゲーションライブラリシークエンスがインプットデータとして必要です。
(ゲノムアセンブリ受託サービス) Dovetail 近接ライゲーションライブラリのリードペアはどのようにスキャフォルディングに使用しますか?
HiRise は、ドラフトアセンブリ上に推定される、偽結合箇所を切り離し、近接ライゲーションライブラリのリードペアで再結合し、より大きなサイズのスキャフォルド配列を作成することができます。そのためにHiRiseはプロジェクトごとに、作成した近接ライゲーションライブラリのインサートサイズ分布モデルを作成します。このモデルは、最初にマップした少量のシークエンスから作成されます。その後、そのモデルの推定値から、どの場所が切り離され、どの場所が再結合されるかを決定します。近接ライゲーションライブラリでは、2つのリードペア間距離が短いものは頻度が多く、長い距離をスパンするものは頻度が少なくなります。 HiRiseは多数の短いリードペアと少数の長いリードペアを使用して、アセンブリのコンティグまたはスキャフォルドを、様々な異なる向きと順序をテストして、その中で、想定されるインサートサイズ分布に最も適した向きと順序をそろえてつないだスキャフォルドを作成します。
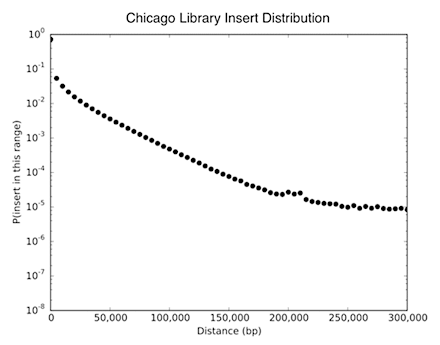
このグラフは典型的なChicagoライブラリのインサート距離の分布を示します。HiRiseによって導き出されたスキャフォルドの向きと順番が正しければ正しいほど、2本のスキャフォルドをつなぐリードペアの距離が、このインサート距離の分布モデルに一致するようになります。
(ゲノムアセンブリ受託サービス) Dovetail メイトペアやフォスミド、BACライブラリなどからのシークエンスデータを持っている場合、これらのデータをHiRiseに使用できますか?
これらの追加データは、現在のところHiRiseパイプラインに直接使用することはできません。しかしこれら追加データは、ドラフトアセンブリの結果を良くする可能性があり、ゲノムアセンブリ全体に見れば貢献します。また、オプティカルマッピングデータなどをお持ちの場合、HiRise後のスキャフォルド配列を改善するのに役に立つ可能性があります。さらに、カバレッジの低いBAC-endシークエンスや、RNA-seqデータなども、最終アセンブリ結果のバリデーションに用いることができると考えられます。
(ゲノムアセンブリ受託サービス) Dovetail 性染色体に興味がある場合は気をつけることは?
性染色体の配列、特に性染色体上の遺伝子を解析したい場合、2x以上のカバレッジが必要になります。
(ゲノムアセンブリ受託サービス) Dovetail ハプロタイプを分けて解析を行うことはできますか?
フェージング済みのドラフトアセンブリを用いることで、ハプロタイプごとのアセンブリを行なうことが可能ですが、その場合にはハプロタイプごとにHiRise解析費用が必要となります。
(ゲノムアセンブリ受託サービス)Dovetail ゲノムアセンブリの結果を良くするためにはどうすれば良いでしょうか?
ゲノムアセンブリの結果には複数の要因が複雑に関係しますが、HiRiseによるアセンブリの結果に最も影響が大きいものの一つは、インプットに使われるドラフトアセンブリのクオリティです。ドラフトアセンブリのコンティグ配列が長く、精度が高いほど、最終のゲノムアセンブリ結果も良くなります。
また、ドラフトアセンブリのクオリティは、リピートやヘテロ接合性、サイズなど、ゲノムの特徴に大きく依存します。またNGSリードの量や種類、使用したアセンブリアルゴリズムにもアセンブリ結果は大きく影響を受けます。
HiRiseアセンブリは、近接ライゲーションライブラリの配列データにも左右されます。近接ライゲーションデータで特にアセンブリに影響が大きいのは、ライブラリのゲノムカバレッジ均一性、リードペア間の距離の分布、シグナルノイズ比です。Dovetail社では、これらが最適な値を示すようなライブラリ調製を行なっています。近接ライゲーションライブラリ作成においては、ほとんどの要因はプロトコルで既に最適化されていますので、サンプルからどれだけ長くてきれいなDNAを抽出できるかが最も影響が大きくコントロール可能な要因の一つです。
(ゲノムアセンブリ受託サービス)Dovetail ゲノムアセンブリの結果はどのように評価することができますか?
標準的な参照ゲノム配列がある生物種の場合、アセンブル結果を参照配列と比較することでエラーの種類や頻度を算出できます。これまでDovetail社では、HiRiseアセンブリの精度を高めるため、参照配列がある生物種のアセンブリ結果を評価することで、プロトコルや解析アルゴリズムを最適化してきました。
参照配列が未知の生物種のアセンブリ結果を評価するためには、ゲノムアセンブリ結果と、進化的に最も近く既知のゲノム配列との間で、シンテニー(染色体上の遺伝子座の並び・順番)を比較する方法があります。もし近縁種で既知のゲノム配列が無い場合には、BAC-ends配列やトランスクリプトームデータを利用することが考えられます。BAC-endデータは数十kbから数百kbに及ぶ長鎖配列です。これらのデータをHiRiseアセンブリの結果配列にアラインすることで、予想インサートサイズやリードの向きを精査し、エラー頻度を算出することに用いることができます。
(ゲノムアセンブリ受託サービス)Dovetail TAD解析を行なうためにはどのようなゲノムアセンブリ結果が必要ですか?
Dovetail社では、以下のような条件を満たすゲノムアセンブリについて、TAD解析を行なう事が可能です。
・近接ライゲーションライブラリのリードペアの40%が1 Kbp以上の距離
・ゲノムサイズ < 10 Gb
・スキャフォルドサイズ < 400 Mbp
・ゲノムの40%以上がユニークにマッピング可能
・1Gbあたり100Mリードペアのカバレッジ